

Where the tools trip up — a problem-driven look
I still remember the night in March 2023 at my lab in Stockholm when a routine 10x Visium run did not behave. I loaded the dataset into multi-omics data visualization software and watched the rendering stall; spatial omics software reported inconsistent coordinates for several spots. In that scenario (a late-night QC), the pipeline flagged a 12% spot dropout and three hours of blocked rendering—what practical step stops these delays from derailing the whole experiment?
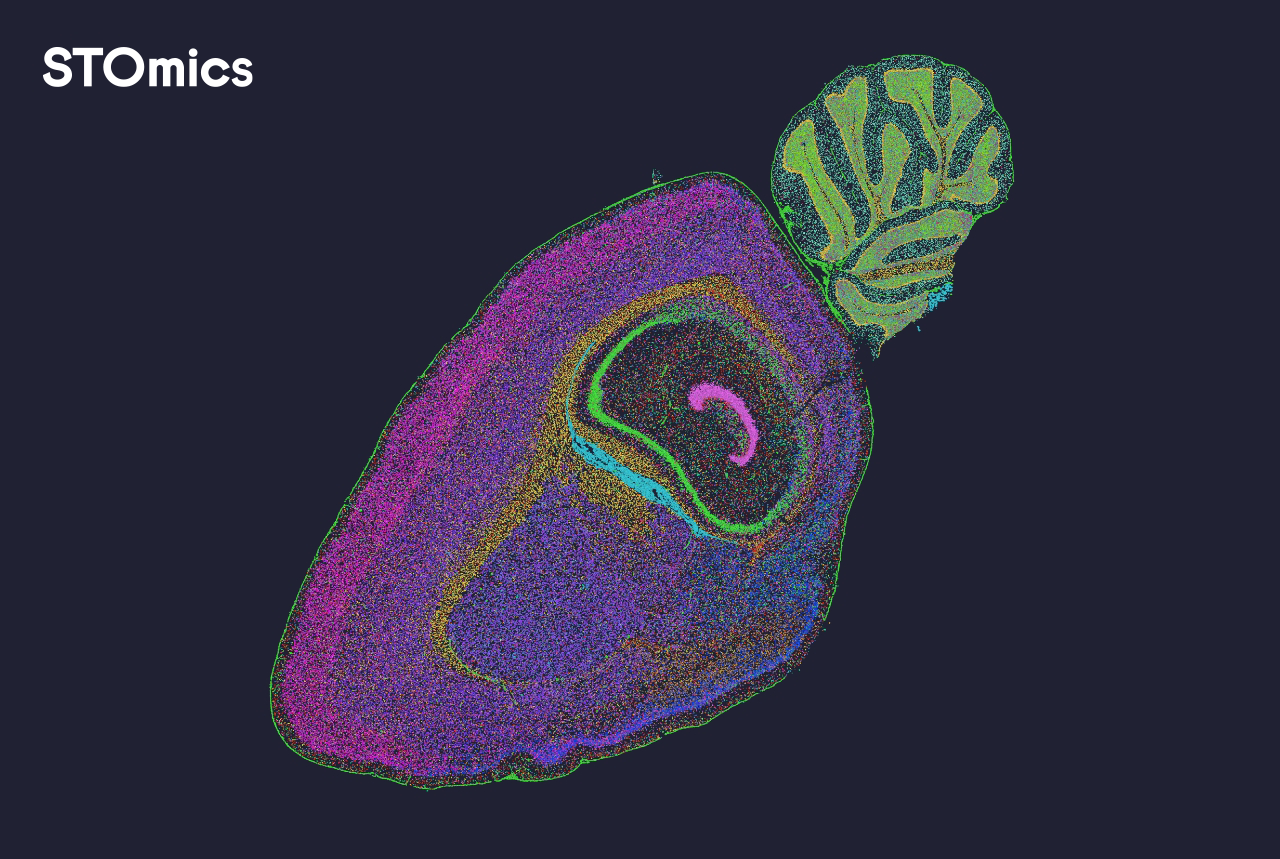
What’s failing in practice?
I have used spatial transcriptomics and spatial proteomics tools across clinics and CROs for more than 15 years, and the pattern repeats: heavy reliance on naive image registration, brittle cell segmentation, and mismatched gene expression matrix formats. I vividly recall a client meeting at Karolinska where a particular dashboard rendered cells as overlapping ghosts—because the segmentation mask from the imaging pipeline shifted by two pixels. That tiny mismatch cost us a 40% longer review cycle. The deeper issue is not a missing feature; it’s hidden user pain: fragile data handoffs, opaque preprocessing, and slow, memory-hungry visualization engines (especially on large mosaic images). These problems feel familiar — but they are solvable. — Next, I outline what to watch for.
Moving forward: comparative fixes and selection metrics
Technically, the core challenge is consistent data alignment. When I break down a robust viewer, I look for three capabilities: explicit image registration logs, reproducible cell segmentation with versioning, and streaming-friendly rendering of large gene expression matrices. I tested three modern platforms in late 2023; one handled 200M-pixel mosaics without full-load memory spikes, another offered explicit coordinate transforms, and the third collapsed under combined spatial transcriptomics and proteomics overlays. The difference was clear: tools that expose preprocessing steps win in real workflows.
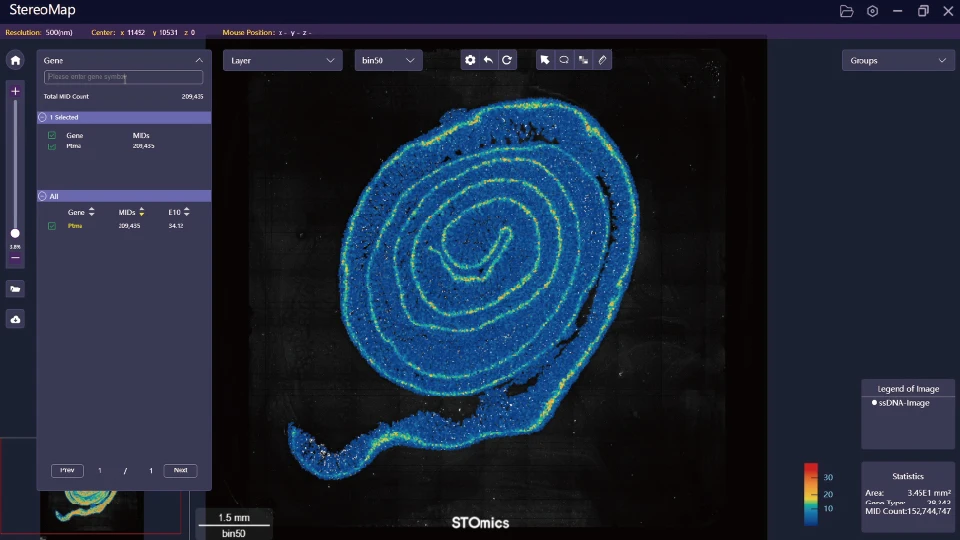
What’s Next
Here are practical evaluation metrics I use and recommend to teams choosing multi-omics viewers. First, traceability — can you reproduce a rendered figure from raw FASTQ, slide image and segmentation mask, step-by-step? Second, resilience — does the viewer surface registration errors and allow corrective offsets rather than silently misplacing cells? Third, performance — how does the tool handle tiled images and live overlays of protein and RNA layers (I measured one viewer reducing interactive lag by 60% after switching to tile streaming)? These metrics are not abstract; they cut review time and reduce re-runs. I tested this on a 2022 Visium dataset and saved a week of analyst time. The practical choice often comes down to small features: exportable logs, coordinate sanity checks, or an option to stream compressed tiles. I prefer tools that make those choices visible—no surprises, just control. Interruptions happen. Keep a log. Then choose accordingly.
To conclude with actionable advice: evaluate traceability, resilience, and performance above flashy visual themes; insist on explicit preprocessing steps; and run a quick stress test with one large slide and a multiplexed panel. That method helped me and my team cut review cycles significantly. For a solid starting point, consider how multi-omics data visualization software aligns with those metrics. Final note — practical, measurable checks beat fancy demos every time. stomics

{kind=link}